


{kind=link}
{kind=link}
慢性中性粒细胞白血病的研究进展
慢性中性粒细胞白血病的研究进展
作者简介:吴学宾,硕士,主任医师。
关键词:
慢性中性粒细胞白血病; 基因突变; 骨髓增殖性肿瘤; CSF3R; SETBP1
中图分类号:R557.3
慢性中性粒细胞白血病(chronic neutrophilic leukemia, CNL)是一种非常罕见的骨髓增殖性肿瘤(myeloproliferative neoplasm, MPN)。由于此前对该病缺乏足够、统一的认识和切实有效的治疗措施, 对其预后并不乐观。近年来, 对本病的研究取得许多新的进展, 深入了解这些进展, 将有助于我们更加深刻地认识本病和指导临床实践。
1 CNL的流行病学
本病1920年由Tuohy E首次报道, 此后对本病一直存在争议, 直至2001年WHO确认诊断分型, 将其归入骨髓增殖性疾病(肿瘤)。期间共报道约150例患者, 2008年WHO重新修订了本病的诊断分型标准, 按新修订的标准判断, 实际诊断的CNL仅约40余例, 临床拟诊的CNL患者最终能够确定诊断仅约51%[1, 2, 3], 且老年人发病率高, 中位发病年龄为66.5岁(15~86岁), 男性略多于女性(1∶ 0.7)。本病预后不良, 生存期短, 中位生存期仅23.5个月(1~106个月), 向急性髓系白血病转化的中位时间是21个月(3~94个月)。最常见的直接死亡原因是颅内出血、疾病进展(或原始细胞转化), 以及化疗或移植相关的毒副反应[1, 4, 5]。我国2007年第3版《血液病诊断及疗效标准》未收录该病, 也说明学界对该病的认识并未统一。
2 CNL的既往诊断标准和鉴别诊断
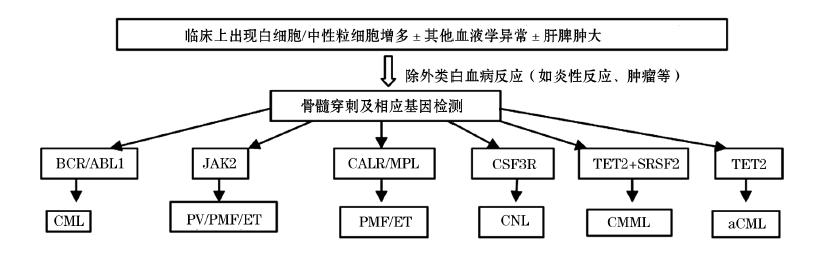
2008年, WHO修订了关于CNL的诊断标准, 虽然增加了一些分子生物学指标, 但仍然是一个除外性的诊断标准, 在鉴别诊断中最主要的是要与不典型慢性粒细胞白血病(atypical chronic myeloid leukemia, aCML)和慢性粒单核细胞白血病(chronic myelomonocytic leukemia, CMML)鉴别[1, 2, 6]。另外, 也需要与类白血病反应(包括炎性反应、感染、肿瘤等)及其他特殊类型的疾病, 如中性粒细胞慢性髓细胞白血病(CML-N)、与浆细胞紊乱相关的中性粒细胞增多症等鉴别[7, 8]。
2.1 CNL与aCML和CMML的鉴别
3种疾病均无Ph染色体或BCR/ABL1融合基因, 无PDGFRA, PDGFRB, 或FGFR1等基因重组, 除CMML规定其外周血单核细胞绝对值> 1× 109/L外, 3种疾病的诊断和鉴别诊断既无分子生物学的标志也无特征性的诊断指标, 主要基于临床细胞学分类检查, 以及除外其他MPN, 如真性红细胞增多症(PV)、特发性血小板增多症(ET)、原发性骨髓纤维化(PMF), 以及MDS或MDS/MPN。
3 CNL的骨髓形态学
CNL的骨髓检查提示, 显著的髓系细胞增殖, 粒红比甚至超过20∶ 1, 中晚幼粒细胞和杆状核细胞增生, 原始粒细胞和早幼粒细胞并不增加, 与CML不同的是嗜碱和嗜酸粒细胞常常缺如; 幼稚红细胞相对减少, 但各阶段分化成熟正常; 巨核细胞数量和形态正常, 也有轻度增高者。按2008年WHO的诊断标准, CNL不存在有意义的细胞形态学异常; 如果存在, 要排除aCML[7]。然而, 也有关于CNL存在细胞形态学异常的报道, 且认为该形态学异常与患者预后相关[11, 12]。骨髓活检无骨髓纤维化的证据, 但也有报道CNL存在严重的骨髓纤维化, 该骨髓纤维化缺乏原发性骨髓纤维化的证据, 是CNL疾病进展至终末期的表现[11], 说明CNL也和其他MPN一样具有向MF转化的特征。
4 CNL的细胞遗传学检查
大部分CNL患者的细胞遗传学检查正常。在Elliott等[1, 13]研究中, 23%的患者有细胞遗传学的异常。常见的细胞遗传学改变包括del(20q)、+21、+8、+9, del(11q), del(12p)和X染色体的异常等[2、11, 12, 13, 14, 15, 16]。这些细胞遗传学的异常对确定诊断无特异性。
5 CNL的分子生物学
WHO在既往分子生物学的检查中对CNL的诊断主要是除外其他的恶性血液肿瘤性疾病[6、17], 如CML的BCR/ABL1、与嗜酸细胞增多和其他异常的髓系和淋巴系统肿瘤的基因改变(PDGFRA、PDGFRB或FGFR1)等均为阴性。近年来, 在分子生物学方面研究的进展, 对诊断CNL提供了极具价值的手段。
5.1 JAK2V617F突变
JAK酪氨酸激酶在细胞因子介导的造血细胞信号通道中发挥着极为重要的作用, 促红细胞生成素、促血小板生成素以及缺乏磷酸化活性的克隆刺激因子3(CSF3)的细胞因子受体通过与其配体结合诱导JAKs的磷酸化, 并进一步下调如STAT通道的转录过程[18]。体细胞的JAK2V617F突变是经典的BCR/ABL1突变阴性的MPN最普遍的突变, PV为95%, ET为55%, PMF为65%, 非经典的MPN不足20%, CMML为8%, MDS和AML罕见[2、19, 20, 21]。在CNL患者中, JAK2V617F的突变为5%~20%[5、22、23], 且特异性也较差[24], 这些研究结果表明, JAK2V617F突变对诊断CNL的价值非常有限。
5.2 CSF3R突变
CSF3R基因(granulocyte colony-stimulating factor receptor, G-CSFR)是造血细胞受体超家族成员之一, 作为G-CSF的受体, 定位于染色体1p34.3, 具有促进中性粒细胞的增殖、存活及分化等功能, 虽然其本身没有内源性酪氨酸激酶活性, 但是可以通过配体结合而改变构象从而刺激多种与其细胞活动范围相关的酪氨酸激酶, 包括JAKs、SRC激酶家族及SYK、TNK等, 重要的通道系统包括信号传导转录STAT、磷酸肌醇激酶PI3-K-AKT以及RAS-MAPK 等[3、25、26], 可见其作用机制非常复杂。作为最新的研究成果, CSF3R的突变与许多疾病相关, 绝大部分与髓系系统疾病相关, 如遗传性中性粒细胞减少症(severe congenital neutropenia, SCN)、MDS、AML及CNL等。基因编码的G-CSFR有多种突变, 目前已知CSF3R的突变至少涉及18种以上, 不同的突变模式与不同的疾病相关, 如p.Thr595Ile(p.Thr618Ile)的突变与CNL相关[25]。
Maxson等[27]在研究恶性血液肿瘤CSF3R的表达时发现, CSF3R突变在27例CNL或aCML患者中有16例(59%)出现; 292例AML中有3例(1%), 8例T细胞和41例B细胞ALL均阴性。Gotlib 等[5]的研究表明, 8/9(89%)的CNL和8/20(40%)的aCML患者被检测出CSF3R突变基因。Pardanani等[26]将临床诊断的CNL35例, aCML19例, CMML94例和PMF76例检测CSF3R突变基因, 结果在13个病例中检测出14例CSF3R突变基因。再按照WHO的标准对这些病例确定诊断, 结果是符合WHO诊断标准的CNL12例(与单克隆球蛋白和淋巴肿瘤无关), 单克隆球蛋白(MG)相关的CNL6例, 可疑CNL但不符合WHO标准的17例; 符合WHO标准的aCML9例, 不符合WHO标准的aCML10例(其中确定为BCR/ABL1阳性的CML2例, CMML3例, MDS/MPN-U2例, PMF1例, MPN-U1例, 系统性组织细胞相关的MDS/MPN-U1例); 另外的共170例CMML和PMF均符合WHO诊断标准。13个病例中的14例次CSF3R突变为12例WHO诊断标准的CNL和1例未定性CNL患者, 所有MG相关的CNL、aCML、CMML和PMF均为阴性结果。CSF3R突变在CNL中的阳性率为100%, 其中CSF3RT618I最为常见占10例, 均符合WHO诊断标准的CNL(占符合WHO标准CNL的83%), 另外3例表现为CSF3RI598I和(或)CSF3RM696T突变。这些结果表明, CSF3RT618I突变基因是CNL的一个高度特异和敏感的分子生物学标记。
5.3 SETBP1突变
SETBP1以170 ku的核蛋白与SET交联, 是肿瘤抑制蛋白磷酸激酶2A(PP2A)的一个负性调节因子, 可以保护SET被蛋白酶的剪切, 使其数量增加从而抑制PP2A的活性[5, 28]。Sení n等[29]对7例患者进行研究, 其中aCML和MPN-U各3例, 1例CNL, 结果显示, CNL表达CSFR3 (T618I), SETBP1 (G870S) 和SRSF2 (P95H); 2例MPN-U表达SETBP1, 其中1例还有SRSF2 (P95H) 和 ASXL1 (E635fs)的表达; 3例aCML患者均未表达SETBP1或CSF3R。Makishima等[28]的一项对于SETBP1的国际合作研究, 在全部727例中共检测出SETBP1基因52例, 占7.2%, 其结果显示, 与SETBP1突变具有显著相关的因素和疾病年龄、-7/del(7q)、sAML(继发性急性髓性白血病)、CMML, 而在原发性AML(pAML)中发生频率很低; 多因素临床资料分析显示, SETBP1突变是一个独立的不良预后因素。Meggendorfer等[30]检查130例MPN和MDS/MPN患者的SETBP1基因, 检出率分别为3.8%和9.4%, 其中以aCML最高为19/60(31.7%), 而且该基因突变与染色体-7和i17(q10)相关。Piazza等[31]检测SETBP1突变的表达, 结果显示aCML为24.3%(17/70), MDS/MPN-U为10%(3/30), CMML为4%(3/82), 4例CNL仅1例表达(25%), 而458例其他的恶性血液肿瘤和344个表达淋巴瘤和其他非造血恶性肿瘤细胞系均表达阴性。临床资料显示, SETBP1突变病例与白细胞增高和不良预后显著相关, 表达SETBP1的aCML中位生存期显著低于未表达SETBP1的aCML患者(22个月vs77个月)[31]。Pardanani等[26]检测SETBP1在符合WHO诊断标准的CNL、aCML、 CMML和PMF患者中, 其SETBP1表达率分别为33%, 0, 7%和3%; 在临床诊断的CNL中, 6例有SETBP1突变, 29例SETBP1阴性, 其中位生存期分别为15个月和26个月。结合临床资料分析认为, 对于CNL患者, CSF3R突变与临床存活无关(P=0.83), 而SETBP1突变可显著缩短患者生存时间(P=0.01)。
6 CNL的新诊断策略和最新诊断标准
通过对基因突变的检测来探索CNL诊断的分子生物学标记, 近年来取得了较大的成就, 使CNL再也不会成为一个仅需要通过用排除性方法来诊断的疾病[35]。Meggendorfer等[36]对近年来有关MPN, 特别是关于CNL、aCML和CMML之间的分子生物学标记特征进行荟萃分析研究, 发现不同的突变基因在不同疾病上的表达具有显著的差异(表1), 通过这些突变基因的检测, 可以对包括CNL在内的有关MPN进行准确诊断和鉴别诊断而不再仅仅拘泥于临床表现和除外性诊断鉴别。
| 表1 六种主要突变基因在CNL、aCML和CMML中的表达及其特异性 |
6.2 诊断最新标准
学者们提出了如下最新的WHO诊断标准修改建议[38]。(1)主要标准:①外周血白细胞≥ 13× 109/L; ②中性分叶核和杆状核细胞> 80%; ③存在CSF3RT618I或其他的CSF3R近膜区突变。(2)次要标准:①骨髓增生显著, 增生以不伴有明显核左移的粒细胞增生为主, 且无病态的粒细胞; ②外周血中, 幼稚粒细胞< 10%, 原始粒细胞< 2%, 且不伴有病态的粒细胞, 单核细胞绝对值≤ 1× 109/L(或单核细胞< 10%); ③存在有一个克隆的标记, 或缺乏反应性/继发性中性粒细胞增多的证据, 包括浆细胞增殖性疾病; ④缺乏BCR/ABL1; ⑤不存在符合WHO诊断标准的其他骨髓增殖性肿瘤。满足所有3个主要标准或至少2个主要标准加所有次要标准才能确诊。
7 CNL的治疗
目前, 本病尚无特异有效的治疗手段, 探索最佳的治疗手段仍然是一项艰巨的任务。
7.1 常规治疗
羟基脲是最基础常规的用药, 主要是降低过高的白细胞或控制脾大[1、5、27]。α 干扰素也是常用的治疗措施, 部分患者也能够获得良好的效果[5、23]。包括沙利度胺、伊马替尼、美罗华等也可用于本病的治疗[1, 5、41, 42, 43, 44]。诱导化疗常用于急变期患者, 但疗效仍不尽人意[41, 42]。异基因造血干细胞移植有望根治本病, 但目前尚无成规模的、有系统的、有效且成功的病例报道[39、45]。
7.2 最新治疗措施
目前主要集中在对于新的JAK-STAT和SRC激酶抑制药磷酸鲁索替尼(Ruxolitinib)和达沙替尼(Dasatinib)的研究, 且已经有报道获得一定疗效。磷酸鲁索替尼和达沙替尼分别对由近膜区突变激活的JAK-STAT通道和由截断性突变激活的SRC家族无受体酪氨酸激酶通道作用敏感[1、3、27、38](图2), 尤其是Ruxolitinib(磷酸鲁索替尼)是FDA近期批准的首个用于治疗高危的骨髓纤维化和真性红细胞增多症的药物, 有学者用该药治疗CNL取得良好效果。对于磷酸鲁索替尼的安全性和有效性的多中心的Ⅱ 期临床试验正在注册进行中, 且正在申请FDA批准用于CNL和aCML的治疗[3、27、38]。这些新的治疗手段将有望真正改变CNL患者的预后。
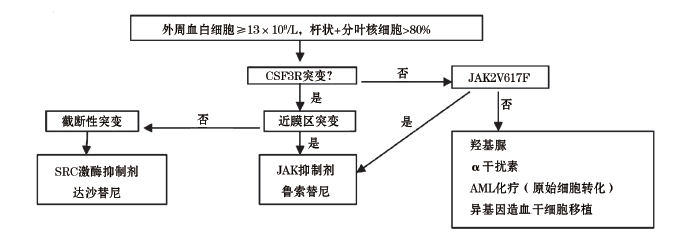 | 图2 CNL的诊断和治疗简易流程 |
CNL是一种罕见疾病, 目前尚有许多不明之处, 尤其对于本病的诊断尚存在不同的看法, 临床治疗也缺乏有效性。新的特异的分子生物学标记的确认, 特别是CSF3R和SETBP1的研究成果, 有望成为诊断CNL和判断CNL预后的重要的分子标记物, 也必将有助于学界形成共识, 为促进本病的治疗开辟新的途径。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|