
{kind=link}
蛋白激酶在大鼠缺血及再灌注脑损伤中的调节作用
引用本文
许忠, 何晓飞, 徐凯, 徐如祥. 蛋白激酶在大鼠缺血及再灌注脑损伤中的调节作用[J]. 武警医学, 2018,29(3): 250-252
XU Zhong, HE Xiaofei, XU Kai, XU Ruxiang. Modulation of protein kinase during ischemia/reperfusion brain injury[J]. Medical Journal of the Chinese People's Armed Police Forces, 2018,29(3): 250-252
Permissions
XU Zhong, HE Xiaofei, XU Kai, XU Ruxiang. Modulation of protein kinase during ischemia/reperfusion brain injury[J]. Medical Journal of the Chinese People's Armed Police Forces, 2018,29(3): 250-252
Copyright©2018, 《武警医学》编辑部
蛋白激酶在大鼠缺血及再灌注脑损伤中的调节作用
作者简介:许 忠,博士,副主任医师。
摘要
目的 探讨蛋白激酶在大鼠缺血及再灌注脑损伤中的调节作用。方法 孕18 d SD大鼠,取胚胎大鼠海马并分离培养海马神经元,加阿糖胞苷抑制神经胶质细胞增殖以纯化神经元,随机分为对照组与实验组,对照组正常培养,实验组缺血时间设定为30 min与60 min,采用Western blot检测蛋白激酶C活性及蛋白表达。结果 对照组、实验组缺血30 min和缺血60 min神经元胞浆PKC活性分别为(6.24±0.27)pmol/(min·mg)、(3.26±0.21)pmol/(min·mg)和(3.05±0.17)pmol/(min·mg),胞膜PKC活性为(2.63±0.13)pmol/(min·mg)、(8.85±0.32)pmol/(min·mg)和(10.63±0.35)pmol/(min·mg),缺血神经元胞浆PKC活性较正常明显下降,胞膜PKC活性明显增加;缺血再灌注损伤后,其胞浆PKC活性分别为(0.97±0.19)pmol/(min·mg)和(0.82±0.16)pmol/(min·mg),胞膜PKC活性为(12.38±0.39)pmol/(min·mg)和(12.66±0.99)pmol/(min·mg),上述改变依然存在并较前者明显。PKCα表达亦呈现上述相似的改变。所有改变均随着缺血时间的延长而加重,与对照组比较,差异有统计学意义( P<0.05)。结论 缺血损伤与PKC的移位激活密切相关,缺血损伤所致的Ca2+超载为其中心环节。
关键词:
脑缺血; 再灌注损伤; 蛋白激酶
中图分类号:R743
Modulation of protein kinase during ischemia/reperfusion brain injury
Abstract
Objective To explore the modulation of protein kinase during ischemia/reperfusion brain injury.Methods Primary hippocampal cultures were prepared from day-18 SD rat embryos. Hippocampal neurons were dissociated by incubation in typsin and purified with arabinosylcytosin (Ara-c), which could inhibit the proliferation of neuroglia. The postischemia time was simplified to 30 min and 60min. Changes of PKC activity in plasma and membrane were assessed by phosphoryl transfer pieces and the expression of PKCα protein was measured by Western blot.Results The activities of cytosolic PKC of the control group, ischemia 30 min group and ischemia 60 min group were (6.24±0.27) pmol/(min·mg), (3.26±0.21) pmol/(min·mg) and (3.05±0.17) pmol/(min·mg)respectively, while the activities of membrane PKC were (2.63±0.13) pmol/(min·mg), (8.85±0.32) pmol/(min·mg)and(10.63±0.35) pmol/(min·mg)respectively. After ischemia/reperfusion brain injury , the activities of cytosolic PKC were (0.97±0.19) pmol/(min·mg)and (0.82±0.16) pmol/(min·mg)respectively, while the activities of membrane PKC were (12.38±0.39) pmol/(min·mg)and (12.66±0.99) pmol/(min·mg)respectively. Ischemia and reperfusion injury significantly increased the activity of membrane PKC and decreased that of cytosolic PKC. These changes became more significant with the extension of ischmia duration. Similar results were also observed in the expression of PKCα protein.Conclusions Ischemia reperfusion injury of rats’ hippocampal neurons results in translocational activation of PKC, especially PKCα, and the activation might damage the neurons by promoting calcium overload.
Keyword:
ischemia brain injury; reperfusion injury; protein kinase
脑缺血或创伤后兴奋性氨基酸过度释放和激活, 兴奋性毒性作用及其所致的细胞内Ca2+超载是引起神经细胞延迟性凋亡, 并导致继发性脑损害的重要原因[1, 2]。越来越多的研究发现, AMPA受体的GluR2亚单位在阻断Ca2+通透过程中起着关键作用[3]。迄今, 有关AMPA受体GluR2亚单位的研究已很多, 但仍有诸多机制需要深入探讨, 如:胞膜表面功能性GluR2亚单位数目是如何改变的?是否受GluR2蛋白总量或代谢过程调节?AMPA受体其他亚单位如何变化?尤为需要关注的是, 上述改变与蛋白磷酸化之间存在何种联系。本研究旨在借助Western blot检测技术探讨蛋白激酶的调节作用, 为深入阐明缺血脑损害发生的分子机制提供理论依据。
1 材料与方法
1.1 海马神经元培养
取孕18 d SD大鼠, 断头处死, 采用体积分数为0.75的乙醇消毒腹部, 取出胚胎, 剪取胎脑, 解剖显微镜下分离双侧海马, 冰D-Hank液洗2次, 剪碎, 质量浓度为1.25 g/L胰蛋白酶37 ℃消化15 min, 血清中止消化, 离心800 r/min, 10 min, 弃上清, 加种植培养液(含质量浓度为200 g/L FCS的DMEM)4 ml, 用吸管轻柔吹打, 筛网过滤, 以1× 106/L细胞数种植于塑料培养皿中(培养皿预置经0.1 g/L多聚赖氨酸处理的盖玻片), 每个培养皿加培养液2 ml, 37 ℃、体积分数为0.05 CO2孵箱内培养。第3天, 在培养液中加入Ara-c10 μ mol/L, 24 h后更换新的培养液(含NGF 25 μ g/L), 以后每隔3~4 d更换一次培养液。
1.2 模拟缺血损伤
体外培养12 d, 从CO2培养箱中取出培养皿, 生物安全柜内吸去培养液, 添加预先去氧的无糖细胞外液(ECS-OGD), 主要成分为116 mmol/L NaCl, 5.4 mmol/L KCl, 0.8 mmol/L MgSO4, 1.0 mmol/L NaH2PO4, 1.8 mmol/L CaCl2, 36 mmol/L NaHCO3, 33 mmol/L Sucrose, 迅速置入专用缺氧培养箱内缺氧30 min和60 min, 缺氧箱内温度为37 ℃, 气体成分的体积分数分别为:0.85 N2, 0.10 H2, 0.05 CO2。分别于缺血后30 min和60 min取出恢复正常细胞外液培养24 h。
1.3 脑缺血损伤致PKC移位激活
(1)胞浆与胞膜蛋白提取:分别取正常培养(对照组)及模拟缺血培养大鼠海马神经元(实验组), 离心, 600 r/min, 10 min, 用D-Hank液洗涤1次后, 离心去上清, 加入1 ml匀浆液[20 mmol/L Tris · Cl, 0.25 mol/L Sucrose, 10.0 mmol/L EGTA, 2.0 mmol/L EDTA, 20 μ g/ml Leupeptin(亮抑酶肽), 1 mmol/L PMSF , pH 7.5]制作匀浆, 4 ℃离心, 100 kg× 60 min, 上清液即为粗提的胞浆提取物, 沉淀加匀浆液(含1 % TritonX 100)4 ℃搅拌30 min; 超速4 ℃离心, 100 kg× 60 min, 上清液即为粗提胞膜成分。(2)PKC活性测定:用中国协和医科大学制的PKC活性检测试剂盒, PKC活性的计算:用1 μ l(γ -32P)-ATP做总计数值(Bq), 用以下公式计算PKC的活性。PKC活性=(反应管Bq-对照管Bq)× 100/(Bq)× 样品蛋白含量(mg)。(3)PKC提取物蛋白含量测定:取PKC提取物0.5 ml, 紫外分光光度计用280和260 nm波长测光密度值(Dλ ), 根据公式(1.45× D280 nm-0.74× D260 nm)× 稀释倍数, 可测得蛋白含量(mg)。(4)PKCα 移位激活(western blot):分别取胞膜蛋白100 μ g作8 %SDS-PAGE凝胶电泳, 转移至硝酸纤维素膜上, 7 %的脱脂奶粉-PBST溶液室温封闭1 h。洗膜后分别加入7 %脱脂奶粉-PBST溶液配制的PKCα 单克隆抗体(1∶ 100), 4 ℃过夜孵育, HRP标记的羊抗小鼠IgG杂交, 加入免疫印迹化学发光试剂进行显影, 运用图像分析系统进行吸光度分析, 强度以吸光度与面积的乘积来表示(A× mm2)。
1.4 统计学处理
采用SPSS 10.0统计软件处理数据, 计量数据以
2 结 果
2.1 脑缺血损伤致PKC移位激活
体外模拟缺血可致大鼠海马神经元胞浆PKC活性明显下降, 并随着缺血时间的延长而降低。与此同时, 胞膜PKC活性明显增加, 同样随着时间的延长其活性更高。恢复氧供应24 h, 上述情况依然存在(表1)。
| 表1 体外模拟缺血损伤大鼠海马神经元PKC活性变化(pmol/min· mg; n=10; |
2.2 缺血损伤后PKCα 的变化
体外模拟缺血损伤后, 胞膜PKCα 较正常明显增加, 相对应的, 其胞浆PKCα 较正常减少, 其胞膜PKCα 所占比例亦较正常(图1)。缺血损伤后PKCα 自胞浆向胞膜的转位增加(表2)。
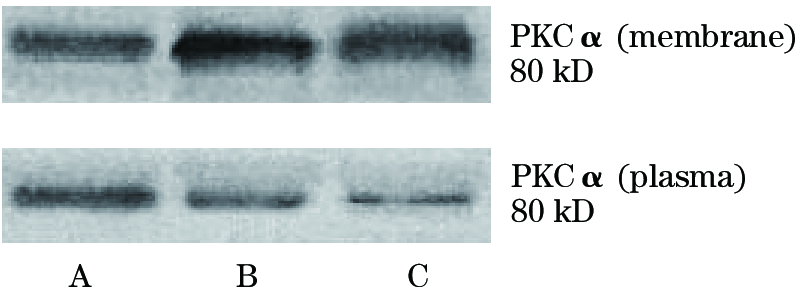 | 图1 缺血损伤致大鼠海马神经元胞膜与胞浆PKCα 变化的western blot结果 A.对照组; B.缺血30 min组; C.缺血60 min组 |
| 表2 体外模拟缺血损伤大鼠海马神经元胞膜和胞浆PKCα 变化(n=10; |
3 讨 论
AMPA受体的磷酸化是该受体发挥作用的主要形式。据文献[4]报道, 多种磷酸化酶都能够使AMPA受体发生磷酸化, 如蛋白激酶A(protein kinase A, PKA)、PKC和钙离子依赖性蛋白激酶Ⅱ (Ca2+/CaM Dependent Protein Kinase Ⅱ , CaMK Ⅱ )等。AMPA受体的所有磷酸化位点均位于C末端的结构域中, 该结构对于受体的磷酸化作用和受体功能的发挥均起着关键作用。GluR2的磷酸化作用主要是由PKC来完成的, 其作用位点在GluR2 C末端的丝氨酸880位点处[5]。
随着技术方法的不断发展, 有关PKC在缺血性脑损伤中的研究得以进一步深入。PKC可分为多种亚型, 如α 、β 、γ 、δ 、ζ 、θ 等[6], 研究表明, PKC的不同亚型对缺血的敏感程度和反应过程是不同的[7]。本研究在显示脑缺血损伤致PKC移位激活的同时, 还发现其移位激活随着缺血损伤程度的加重而愈发明显。进一步采用western blotting技术定量神经元研究缺血损伤后胞内和胞膜上PKCα 含量变化, 结果显示, 胞膜PKCα 含量较对照组显著增加, 而胞浆PKCα 则低于对照组, 提示缺血损伤后PKCα 自胞浆向胞膜的转位增加。至此, 我们验证脑缺血损伤可以导致胞膜PKC活性增加的基础上, 也发现伤后PKC从胞浆到胞膜的移位过程, 结合文献[8], 我们推测, 突触后膜表面AMPA受体GluR2亚单位表达水平和含量变化可能与PKCα 的移位和激活存在某种相关关系。与此同时, 我们还发现, 大鼠海马神经元缺血损伤后, 胞膜PKCα 占细胞PKCα 总含量(胞膜+胞浆)的百分比也高于正常, 随缺血损伤程度的加重, 该比例变化更为显著。
PKC移位激活在脑缺血损伤中作用的可能机制。缺血损伤激活PKCα 后, PKCα 可通过受体结合蛋白与GluR2磷酸化位点(Ser880)结合, 使GluR2在Ser880位点发生磷酸化。由于GluR2的磷酸化位点与谷氨酸受体交互作用蛋白(GRIP)的结合位点是同一位点[9], PKCα 与GluR2的结合及其所致的Ser880位点的磷酸化, 则可减弱GluR2与GRIP的亲和力, 减少突触AMPA受体GluR2亚单位的表达和富集; 同时, PKCα 与GluR2结合二聚体还可阻断GluR2亚单位与ABP和GRIP的结合, 直接影响由ABP/GRIP介导的AMPA受体的突触锚定。上述过程促使GluR2与锚定蛋白解离, 并进而启动胞吞过程, 最终使得功能性AMPA受体GluR2亚单位数量减少。
再者, PKC是一种钙/磷依赖的蛋白激酶, 正常情况下, PKC几乎以无活性的形式存在于胞浆中。缺血脑损伤所致的Ca2+超载, 通过某种特定的机制激活PKC[10]。基于视网膜细胞的研究发现, PKC对通过AMPA受体的Ca2+内流具有调节作用[11]。由此不难看出, AMPA受体、Ca2+超载与PKC在缺血脑损伤中相互影响, 而Ca2+超载为其中心环节。
总之, 我们认为, 缺血损伤后PKC的移位激活直接影响GluR2突触膜表面的锚定与解离, 使膜表面受体含量减少, 在调节突触后膜表面AMPA受体GluR2亚单位的含量及功能上发挥着不可或缺的作用。
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|