




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
利拉鲁肽对大鼠心肌微血管内皮细胞缺氧复氧损伤的影响
[李丹丹, 张颖, 陈韵岱 ]
]
]
|
|


目的 探究胰高血糖素样肽-1(glucagon-like peptide-1,GLP-1)类似物利拉鲁肽(GLP-1 analogue,Liraglutide)对大鼠心肌微血管内皮细胞(cardiac microvascular endothelial cells, CMEC)缺氧复氧损伤的影响。方法 双酶消化法体外分离培养SD大鼠CMEC,实验的细胞分为正常对照组、Liraglutide(0~100 nM)组、H/R组、H/R+Liraglutide组。建立缺氧复氧模型(hypoxia/reoxygenation model,H/R)诱导的细胞氧化应激凋亡模型,流式细胞学检测细胞凋亡率,DCFHDA荧光探针检测胞内ROS的变化免疫荧光染色和Western Blot检测XO、caspase3等蛋白分子的表达情况,通过比较各组中XO、ROS以及凋亡相关蛋白caspase3的表达情况阐明Liraglutide在抗细胞氧化应激过程中的保护作用机制。结果 与正常对照组细胞相比,H/R模型条件诱导细胞中XO表达增加,生成过量ROS及凋亡蛋白的表达,最终导致凋亡细胞数量显著提高至20.66%±1.30%。Liraglutide预处理12 h抑制了H/R诱导的XO的激活,最终降低胞浆过量ROS并将细胞的凋亡数量降低至8.36±1.19%。结论 H/R模型导致XO-ROS激活,生成过量胞浆ROS,最终介导了CMEC的线粒体凋亡通路激活,而予以Liraglutide预处理可以逆转上述过程。
Objective To explore the effects of liraglutide, a GLP-1 analogue, on CMECs and the mechanisms by which liraglutide reduces the oxidative stress apoptosis of CMECs under a hypoxia/reoxygenation (H/R) model.Methods In vitro cultured CMECs of SD rats were purified with the differential adhesion method and identified immunocytochemically using CD31 antibody. GLP-1R and CD31 were assessed by co-location immunohistochemistry. MTT assay was performed to assess the proliferation of the first-generation cells exposed to different concentrations (0-100 nM) of liraglutide. An H/R model was used to induce cellular apoptosis, and the apoptotic rate was assessed by low cytometry. DCFHDA was used to evaluate the ROS contents. Western blot and immunohistochemistry were used to assess the expressions of XO and caspase3. The expressions of XO, ROS and caspase3 in each group were compared to illustrate the liraglutide-mediated anti-apoptotic functions.Results The H/R model induced a higher expression of XO, and consequently generated excessive ROS which was responsible for cellular apoptosis (20.66%±1.30%). Lirglautide pretreatment for 12 h could suppress the XO activation, ROS outburst and cellular apoptosis (8.36%±1.19%).Conclusions This study has confirmed that H/R can induce CMECs’ oxidative damage through the XO-ROS injury signals, and that liraglutide pretreatment may suppress such damage to CMECs.
冠心病心肌梗死已经成为严重危害人类健康的头号杀手, 直接PCI术是目前心血管内科推荐的最为重要的有效治疗手段[1]。但是, 在临床实践中发现, 在解除机械性梗阻恢复血流供应后, 其末端血流供应的心肌组织并没有完全有效的血流灌注, 即出现无复流现象(no reflow)[2, 3]。无复流的发生减少了血运重建带给患者心功能恢复的益处, 使得住院死亡和再发心肌梗死的概率增加4~10倍, 是影响患者即刻和长期预后的重要因素[4, 5, 6, 7, 8]。无复流是指在开通梗死相关动脉时, 微循环血流阻力增大而导致的微循环灌注不良的现象[3]。再灌注时期大量活性氧(reactive oxygen species, ROS)的生成被认为是细胞发生IR损伤的重要启动因素[9, 10, 11]。内皮细胞, 尤其是冠脉末梢的心肌微血管内皮细胞(cardiac microvascular endothelial cells, CMEC), 作为血液和组织之间最重要的屏障, 在缺血再灌注损伤中是最先被累及的, 是组织缺血再灌注后产生ROS的重要来源。内皮细胞中ROS的生成有多种来源, 其主要的来源是NADPH氧化酶、黄嘌呤氧化酶(xanthine oxidase, XO)、线粒体呼吸链、单核巨噬细胞来源等[12]。在多项研究中发现, XO在肝脏内皮细胞、肺动脉内皮细胞以及CMEC中有较高的表达量[13]。并且, XO介导氧化应激损伤以及细胞凋亡在再灌注损伤中可能发挥重要作用[14]。胰高血糖素样肽-1(glucagon-like peptide-1, GLP-1)是一种由肠道内分泌细胞分泌的肠促胰岛素[15, 16, 17]。近年的基础研究发现, Liraglutide除降糖作用以外, 还具心血管保护作用, 可减少心脏血管炎性反应、心肌缺血再灌注损伤、减少心肌梗死面积、抑制心肌细胞凋亡等[18, 19, 20]。本研究通过提取并体外培养SD乳鼠的CMEC, 在H/R模型条件下, 模拟细胞氧化应激损伤过程, 予以Liraglutide预处理12 h对CMEC进行干预, 探究药物的保护作用和机制。
主要药品与试剂 Liraglutide、MTT试剂、Hoechst 33342染料购买于美国, Sigma公司; DCFHDA-ROS探针、DHE-ROS探针、丙二醛脂质过氧化物检测试剂盒、GSH检测试剂盒、超氧化物歧化酶检测试剂盒均购于北京碧云天公司; Annexin V-FITC/PI试剂盒(BD Biosciences, 美国); caspase-3(1∶ 1000, Cell Signaling Technology), Bcl2 (1∶ 2000, Abcam), Bax (1∶ 2000, Abcam), and XO (1∶ 1000, Abcam); CD31抗体(Bioss Inc. 中国); GLP-1R抗体(Bioss Inc. 中国)。5~6 d、质量为12~16 g的SD乳鼠(雌雄不限), 购自解放军总医院动物室, 该研究中动物、细胞来源, 符合国家《实验动物管理条例》及解放军总医院伦理准则。
1.2.1 CMEC的提取和培养 参照Nishida等[21]双酶消化分离原代细胞, 并在此基础上进行了改良。简述如下:5只乳鼠处死后取出左心室, 75%乙醇中浸泡15 s, PBS清洗3遍后剪碎组织块, 0.2%Ⅰ 型胶原酶37 ℃水浴持续震荡搅拌消化10 min, 观察待组织块明显变小后, 加入0.25%胰蛋白酶37 ℃水浴继续消化3 min, 随即终止消化, 200目滤网滤过消化后的液体, 1000 r/min, 7 min离心分离细胞和多余的培养液, 离心完成后弃掉上清液, 以1× 105/ml的密度缓慢吹打细胞后, 37 ℃、5%CO2、饱和湿度培养箱中培养细胞4 h, 去除悬浮未贴壁的细胞, 继续培养24 h后第一次换液, 待细胞至80%融合生长后, 0.25%胰蛋白酶消化传代。根据实验需要使用第一或第二代细胞。CD31抗体进行免疫细胞化学染色鉴定CMEC纯度。缺氧4 h加复氧4 h, 诱导CMEC凋亡模型。
1.2.2 流式细胞术检测 模型建立成功后, 细胞诱导完成时, 胰蛋白酶消化收集细胞, 随后吹打重悬至200 μ l的Annexin V-FITC缓冲液中, 再加入5 μ l的Annexin V-FITC溶液进行37 ℃孵育(避光条件下30 min), 最后再加入10 μ l 的PI与上述液体共同避光条件下孵育5 min, 结束后立即对细胞进行流式细胞学定量分析。
1.2.3 ROS检测 ROS探针作为检测细胞内ROS的方法, 细胞模型建立完成后, 更换为正常培养液, 将10 μ M的DHE加入培养体系中, 随后在37 ℃黑暗中处理30 min, DAPI染核3 min, 随后进行常规PBS洗涤共3次, 后用共聚焦显微镜观察胞内ROS含量变化。在进行ROS定量检测时, 使用流式细胞学方法检测。
1.2.4 MDA、GSH、SOD、GPx检测 酶标仪分析GSH、SOD、MDA、GPx的含量变化, 按试剂盒说明书操作。首先将处理的细胞收集后进行离心沉淀, 破膜后收集细胞蛋白进行ELISA检测。预先绘制标准蛋白含量曲线, 然后按照说明书进行ELISA双夹心法检测, 根据酶标仪测定的吸光值计算最终的蛋白含量及蛋白活性。
1.2.5 免疫荧光染色 细胞处理完成后, 弃掉培养液并使用PBS清洗3遍, 4%冰甲醛常温固定10 min, 0.5% Triton X-100通透细胞膜5 min, 山羊血清10%室温封闭细胞特异性抗原1 h, 一抗孵育(1∶ 250) 4 ℃过夜。PBS漂洗3次后加二抗(1∶ 2000), 室温避光孵育2 h。滴加DAPI, 孵育5 min, 荧光显微镜观察拍照。
1.2.6 干扰RNAi 本实验中针对XO蛋白进行抑制表达实验。siRNA的设计由上海吉玛公司完成, 序列如下:siRNA: 5'-CCACCUCCAAGAUUCAUAUTT-3; Anti-sense: 5-CCGAGAAUGAGCCUGAUUU-3, 阴性对照组的序列如下:siRNActrl: 5-UUCUCCGAACGUGUCACGUTT-3; Anti-sense:5-ACGUGACACGUUCGGAGAATT-3。
1.2.7 Western blot检测 各组处理结束后, 提取总蛋白。10% SDS-聚丙烯酰胺凝胶电泳分离, PVDF膜湿转后山羊血清室温封闭1 h, 加入相应一抗4 ℃孵育过夜, TBST 洗膜后使用HRP 标记的相应二抗(1∶ 3000)室温反应60 min, 暗室曝光, 以β -actin作为内参。采用Quantity One软件采集信号和灰度扫描。实验重复3次。
采用SPSS 17.0软件进行统计, 计量资料采用
培养的CMECs呈梭形, 形似鹅卵石, 贴壁培养24 h后开始生长(图1A), 72~96 h细胞长满瓶底, 呈典型CMEC的铺路石样结构(图1B)。使用免疫荧光技术对CMECs表面标记分子CD31(图1C)与Ⅷ (图1D)因子进行鉴定, 提示分离的CMEC没有被心肌细胞和成纤维细胞污染, 纯度较高, 可以供下一步实验使用。
 | 图1 原代CMEC的培养与鉴定 A-B.CMEC接种后24 h和72 h的细胞形态学观察; C-D. CMEC表面标记物CD31和VIII因子的免疫荧光鉴定 |
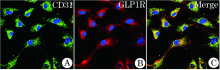
于倒置荧光显微镜下观察, CD31单克隆抗体与GLP-1R多克隆抗体进行共定位染色, 细胞内分别呈绿色和红色荧光, 融合后提示双阳性率> 95%(图2)。
 | 图2 CMEC上GLP-1R的表达情况 A.CD31单克隆抗体定位染色; B.GLP-1R多克隆抗体定位染色; C.融合后 |
同一时间点上OD值随Liraglutide药物浓度增加而显著升高, 于100 nM组达到最高促生长浓度(P< 0.05)。不同时间点上, 总体趋势提示细胞于药物干预24 h后出现对数生长期, 于84 h后进入生长平台期(图3)。12 h细胞密度无明显的统计学差异, 因此将药物预处理12 h作为后续实验的干预条件。
 | 图3 不同浓度Liraglutide对细胞增殖能力的影响 |
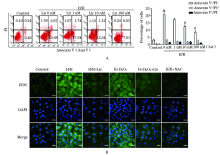
与正常对照组细胞相比, H/R模型导致早期凋亡细胞(Annexin V+/PI-)数量显著提高至20.66%± 1.30%(P< 0.05)(图4A)。而Liraglutide可以显著降低早期细胞凋亡的数量, 并且呈现浓度依赖性(Liraglutide 1 nM 17.38%± 0.82%; 10 nM 12.49%± 1.02%; 100 nM 8.36% ± 1.19%; P< 0.05 vs H/R)。同时发现100 nM Liraglutide对于细胞的保护作用是最强的, 因此后续实验选用100 nM预处理12 h作为干预条件。同时, H/R组细胞内的ROS显著上升, 而Liraglutide干预后可以显著降低胞浆内ROS含量(图4B)。缺氧后给予H2O2可以显著提高胞浆内的ROS, 同时伴随着凋亡蛋白caspase3、Bax等蛋白的显著增加; 而使用ROS清除剂NAC作为阴性对照, 一方面中和了过度的ROS, 另一方面也显著降低了凋亡蛋白的含量(图5)。此外, 在H/R模型下, SOD、GSH和GPx的含量显著下降而MDA在胞浆内大量聚集, 而Liraglutide干预后可以显著上升上述抗氧化因子SOD、GSH和GPx, 同时表现为下降的MDA含量(图6)。
 | 图4 Liraglutide对H/R模型下的CMEC凋亡率的影响 A.H/R模型条件下, Liraglutide对细胞凋亡率的作用; B.DCFHDA探针检测各组间胞浆ROS荧光强度的不同 |
 | 图5 Western Blot检测各组间凋亡蛋白和保护性蛋白表达的差异 A.色谱图; B.定量分析 |
 | 图6 H/R模型以及Liraglutide预处理对细胞MDA、GPX、SOD、GSH含量的影响 |
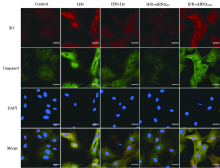
在H/R模型条件下, XO的含量mRNA和蛋白水平均有显著升高(图7A-B)。使用siRNA敲低XO, 可以显著减少H/R模型下ROS的产生(图7C-D)。同时, 过度的XO生成会伴随着caspase3表达的明显提高, 敲低XO则会显著减少caspase3的生成。使用Liraglutide预处理, XO的mRNA水平和蛋白水平均有显著降低。同时, 敲低XO可以进一步降低胞浆内caspase3的表达(图8)。
 | 图7 Liraglutide对XO-ROS的影响 A-B. PCR和Western Blot检测各组间XO表达量的不同; C-D. 流式细胞学定量检测各组间DCFHDA标记的胞浆ROS平均荧光强度的不同 |
 | 图8 Confocol观察不同组间XO和Caspase3荧光共定位染色 |
微血管是终末循环末梢, 在很大程度上决定组织灌注水平和氧供平衡。缺血后的再灌注时期产生了大量的活性氧ROS, 进而诱导细胞内过度的氧化应激水平, 损伤细胞内蛋白质、内质网、线粒体等亚细胞结构的功能, 从而导致了细胞的功能障碍以及随后的细胞程序性死亡过程[22]。而值得注意的是, 在心脏的组织细胞中, 心肌微循环内皮细胞是最易受到攻击的, 由于其在第一时间收到缺氧损伤的刺激, 并且在第一时间恢复了血流[10], 因此微循环内皮细胞比心肌细胞和其他组织结构更早发生再灌注损伤, 出现临床上常见的介入治疗后再灌注并发症-无复流。由于微循环的破坏, 使得组织最终的能量和氧供得不到很好的补充, 因此进一步加重心肌梗死面积的扩展以及随后的心功能降低, 造成心肌梗死围术期的病死率显著提高[23]。由此保护介入术后再灌注时期的微循环的稳定, 将会显著提高PCI治疗的效果, 并增加心肌梗死患者围术期的临床获益。
目前研究发现, 再灌注时期, 复氧期将会产生大量的ROS, 诱导细胞内过度的氧化应激, 损伤细胞的功能, 造成微血管内皮细胞的凋亡。细胞内ROS的来源多种多样:线粒体呼吸链[24]、NAD(P)H[25]、XO[26]以及一些蛋白底物代谢过程中产生的负离子电荷。由于内皮细胞中线粒体的缺乏, 因此线粒体介导的ROS产生并不会很大程度上影响细胞的功能[27]。另一方面值得关注的是, XO途径介导的ROS爆发可能是内皮细胞氧化应激损伤中被忽略的一部分。笔者团队的前期研究提示, CMEC中H/R条件下XO表达和转录明显增加[28], 有研究认为XO是微血管内皮中ROS大量产生的主要因素之一[29]。XDH作为XO的前体形式在细胞内是持续表达的, 在H/R条件下, 大量XDH转变为XO[30], 生成过量ROS。其具体的机制为:缺氧条件下, 细胞内的ATP由于大量消耗被分解为ADP、AMP并最后代谢成为次黄嘌呤。一旦进入再灌注时期, 在氧的介导下, XO大量代谢次黄嘌呤生成尿酸的过程中将会生成大量的氧负离子[31], 由此造成细胞内ROS含量的显著升高。因此在我们的实验中, 建立了H/R模型来模拟缺血再灌注损伤。在模型中, 复氧期将会显著提高CMEC中的XO的含量, 从而诱导ROS的爆发。由此说明H/R激活的XO是细胞内ROS含量的重要决定因素, 干预XO的蛋白水平以及转录活性, 将会显著降低细胞内过度的活性氧, 从而抑制氧化应激介导的细胞凋亡。但是XO-ROS是如何诱导细胞凋亡的, 目前研究甚少。
正常情况下, 机体内ROS生成和清除系统处于动态平衡, 由于多种原因导致的ROS生成增多或清除能力下降都会导致细胞的氧化应激损伤。细胞内过量ROS生成可导致细胞脂质过氧化水平增高、DNA氧化损伤、蛋白质过氧化以及通过多种途径诱导细胞凋亡, 例如:激活细胞NF-κ B转核, 激活P53或SAPK通路诱导细胞, 以及线粒体介导的细胞凋亡通路[25]。本实验发现在H/R模型中, 由XO来源的过量胞浆ROS, 是诱导细胞凋亡的关键。进一步实验发现, 予以Liraglutide预处理或敲低XO, 胞浆ROS生成明显减少, 同时可以在一定程度上降低caspase3表达量, 最终保护CMEC在H/R条件下的结构和功能。本实验证实了在H/R条件下, XO-ROS是最终线粒体呼吸链ROS爆发的重要上游激活因子, 应当作为早期保护细胞氧化应激损伤的重要靶点进行更多的研究。
总之, Liraglutide可以通过恢复胞内正常的XO-ROS水平, 从而降低H/R导致的CMEC氧化应激凋亡, 促进细胞存活。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|